成果介绍
铂纳米催化剂常被用于电催化析氢反应。这些纳米结构包含不同的表面位点,包括(111)和(100)面以及它们之间的边缘位点。确定准确的活性位点对于优化催化剂设计至关重要,但仍然具有挑战性。
加州大学洛杉矶分校段镶锋教授、黄昱教授,加州理工学院William A. Goddard III院士等人结合电输运光谱(ETS)和反应力场(ReaxFF)计算,描绘了铂纳米线上的氢吸附,并揭示了两个不同的峰:在0.20 V的峰与(111)和(100)面有关,在0.038 V的峰与边缘位点有关。同时进行的ETS和循环伏安法表明,边缘位点的吸附与HER的开始一致,表明边缘位点的关键作用。ReaxFF分子动力学计算证实,HER在边缘位点的活化势垒较低,转化频率高2到4个数量级。碱性介质中的ETS在边缘位点上的氢吸附受到抑制,导致HER动力学更加缓慢。这些发现解决了铂表面上不同位点的难以捉摸的作用,为HER催化剂的设计提供了关键的见解。
相关工作以《Edge sites dominate the hydrogen evolution reaction on platinum nanocatalysts》为题在《Nature Catalysis》上发表论文。值得注意的是,这也是段镶锋教授、黄昱教授共同在《Nature Catalysis》上发表的第8篇论文。

图文导读

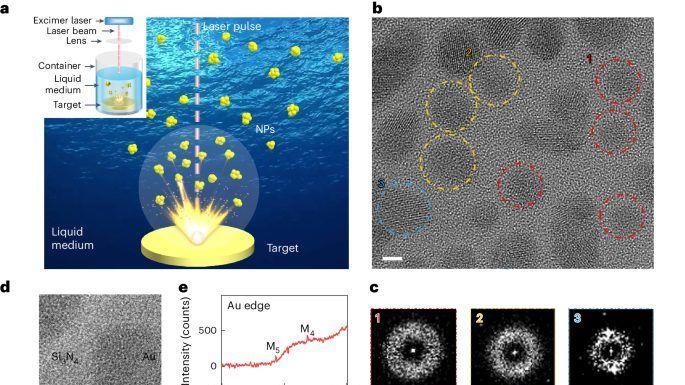
为了确保有效的传质到Pt NW上,专门设计了一个微流体系统,用于有效地将新鲜电解质传输到Pt NW工作电极(图1a)。在实验设置中,使用了两个独立的电测量通道(图1b):(1)在栅极通道中记录直流电流,用于测量电化学电流(即CV);(2)在源极漏极通道中施加交流电,同时监测Pt NW电导率。这两个通道是独立的,彼此完全解耦,允许在存在大法拉第电流的情况下精确测量电导率。 完全活化的PtNWs的平均直径为1.90±0.39 nm(图1d)。图1e显示0.23 nm和0.19 nm的晶格间距,分别对应于Pt的(111)和(200)晶面。这表明PtNW表面由四个(111)和两个(100)面组成,符合平衡结构(图1f),并具有Wulff结构预测的最小表面能。

为了探究表面Had在HER动力学中的作用,重点研究了HUPD和HOPD电位区。在0.1 M HClO4溶液中,在0.05-0.70 V电位范围内测量的CV和电导率变化的典型曲线显示了两个不同区域Pt电极的共同特征(图2a):(1) 0.05-0.40 V发生HUPD,以及(2) 0.40-0.70 V,即所谓的双电层区域。
相应的ETS研究表明,双电层区域的电导率变化很小,但由于表面吸附的水分子逐渐被氢取代,HUPD区域的电导增加更大。这种电导率的增加与氢覆盖表面的电子散射效应比水覆盖表面小的预期是一致的(图2b)。HUPD区域的电导率变化与H的总体覆盖直接相关。因此,电导变化的导数预计与每个电位下的氢吸附电流(即CV)相关。事实上,导函数的变化与氢吸附分支的CV曲线非常相似(图2c)。特别是,在电导率导数结果中,可以清楚地分辨出0.2 V处常见的HUPD峰,证明了ETS电导信号对氢吸附电流的高保真重建。
对比CV和电导率导数曲线,HUPD区域具有高度的一致性(>0.20 V);然而,发现在HER平衡电位附近,CV电流开始偏离导数电导变化。这种差异可以归因于在N2饱和环境下,该电位区域的CV电流中有额外的小HER电流贡献。这样的HER电流不会改变表面吸附质的状态,因此不会产生可测量的ETS信号。为了验证这一假设,分别计算了氢吸附和解吸过程中消耗的电荷和产生的电荷,发现氢吸附过程中消耗的电荷明显大于解吸过程中产生的电荷(图2d),表明氢吸附过程中有额外的HER电流贡献。 重要的是,氢吸附和解吸过程之间的起始电位与CV和ETS之间的差异在很大程度上是一致的(图2c),证实了这种差异的相似起源。简而言之,对于N2饱和溶液中的氢吸附过程,在接近HER平衡电位的电位处会产生少量的H2,这有助于在表面Had之外产生额外的还原电流。相反,在氢解吸过程中,电荷仅来自表面氧化。
通过ETS测量将表面吸附物信号与大HER信号解耦,进而能够在活性HER过程中探测表面吸附物。为此,将电导率测量扫至HER区域,在此区域产生较大的法拉第电流(图2e)。HER区域的CV电流比图2a中的HUPD电流大得多,因此氢吸附的电流响应完全被HER电流掩盖,因此无法通过CV方法直接评估。因此,这个电位区域(例如HOPD)的额外氢吸附一直是难以捉摸的。有趣的是,ETS测量显示电导率持续增加,直到电位为-0.1 V,这表明在该电位区域有额外的表面氢吸附。 通过区分电导率变化,作者发现了两个氢吸附峰,包括在0.20 V处常见的HUPD峰和在0.038 V处新的氢吸附峰(图2f)。考虑到在高于0.05 V电位下吸附的氢通常被指定为HUPD,将这个0.038 V的新峰指定为HOPD。特别有趣的是,HOPD峰值电位与HER电流快速增加的开始时间一致(图2f),这表明HOPD在HER过程中起着关键作用。

总的来说,ReaxFF计算表明,Had覆盖率随着电位的降低而增加。为了探索不同表面位点的氢吸附动力学,在不同电位下进行分子动力学分析(图3)。很明显,在0.158 V时,氢吸附在Pt(111)和Pt(100)位点上,而边缘位点仍未被占据(图3a)。在0.110 V时,氢在Pt(111)和Pt(100)上的覆盖接近完全覆盖(单层的0.99%),并且一些氢原子开始吸附在(111)/(100)边缘和(111)/(111)边缘位点上(图3b)。在接近HOPD的峰值电位0.062 V时,新吸附的Had主要聚集在(111)/(100)和(111)/(111)边缘(图3c)。在0.032 V时,两种类型的边缘位点都接近完全覆盖(图3d),而在-0.052 V时,所有边缘位点都被完全覆盖(图3e)。
总体而言,观察到一个两步式的氢吸附过程,(111)和(100)台阶位点的吸附电位区域与实验HUPD区域一致,边缘位点的吸附电位区域与实验HOPD区域接近。

为了解决氢在不同位点上的吸附过程,在最后0.5 ns中,对每个电位下不同位点上的氢原子总数进行了平均,并绘制了电位(图4a),包括(111)和(100)台阶位点,以及(111)/(100)和(111)/(111)边缘位点。结果表明,氢吸附首先发生在(111)和(100)表面。当这些台阶上的氢吸附接近饱和时,氢原子开始在边缘位点吸附,在-0.1 V左右饱和。
为了估计氢在不同位点的吸附峰位置,对吸附曲线进行了非线性曲线拟合(图4b),并对拟合结果进行了微分(图4c)。H数目的导数与每个电位下的氢吸附电流成正比,显示出明显的氢吸附峰:位于0.218 V处较小的峰对应于Pt(100)面吸附的氢;在0.218 V处较大的峰对应于Pt(111)面吸附的氢;位于0.056 V处较小的峰对应于(111)/(111)边缘吸附的氢;在0.056 V处较大的峰对应于(111)/(100)边缘吸附的氢。 将理论预测的氢吸附曲线与实验ETS信号进行比较,发现实验HOPD峰值明显大于理论峰值(图4d)。将这种差异归因于不同大小的Pt NWs。为此,还研究了不同尺寸的Pt NWs,发现HOPD峰的相对强度随着Pt NW直径的减小而增加(图4d)。其中,1.4 nm Pt NWs与实验值相当。

考虑到HUPD和HOPD的局部成键环境不同,预计它们会表现出非常不同的HER动力学。在平衡电位下,反应通过Volmer-Tafel机制进行,Tafel步骤的速率很大程度上取决于不同氢的重组(图5a、b)。模拟结果表明,HUPD是由中空位点(Pt(100)面上的四倍中空位点和Pt(111)面上的三倍中空位点)吸附的氢产生的,而HOPD是由边缘位点吸附的氢产生的。 基于(111)和(100)台阶空心位点和(111)/(100)和(111)/(111)边缘位点的组合,确定了七种不同的Tafel机制。计算了这七种Tafel机制的最小能量途径(图5c),并确定了相应的自由能活化能(图5d)。 计算表明,仅涉及Pt(111)表面位点((111)H-H)的反应活化能为0.79 eV,这与先前Pt(111)的计算结果一致。有趣的是,(111)/(100)E-E的活化能为0.57 eV, (111)/(111) E-E的活化能为0.61 eV,明显低于其他5种机制的活化能(0.67-0.79 eV)。

在同一器件上测量的CV表明,Pt NWs在-0.10 V电位下,0.1 M KOH溶液中的HER电流比0.1 M HClO4溶液中的HER电流小一个数量级以上(图6a)。与酸性电解质不同的是,在碱性介质中,ETS测量结果显示,在低于0.20 V的电位下,电导率仅略有增加(图6b),表明该电位区域额外的氢吸附有限。相应的电导率变化导数显示,在0.03 V电位处有一个小的HOPD峰值,这也与HER电流的开始时间相吻合(图6c),这表明它在碱性条件下的HER过程中也起着关键作用。
尽管如此,与酸性介质中的HOPD峰值强度相比,碱性介质中的HOPD峰值强度大大减弱(图6d),表明边缘位点的平衡氢覆盖率要低得多。因此,边缘位点HOPD的减少在很大程度上解释了碱性介质中更缓慢的HER动力学。
文献信息
Edge sites dominate the hydrogen evolution reaction on platinum nanocatalysts,Nature Catalysis,2024.