
在美国布朗大学的一项突破性研究中,研究人员在药物发现和理解蛋白质的复杂活动方面取得了关键的飞跃。这一进步,通过创新使用AlphaFold 2来利用人工智能的力量,为预测蛋白质的动态构象(dynamic conformations of proteins)设定了新的前沿。这项研究发表在《自然通讯》期刊上,不仅推动了我们对蛋白质动力学的理解,而且有望加快新疗法的发展。
揭开蛋白质之舞的面纱
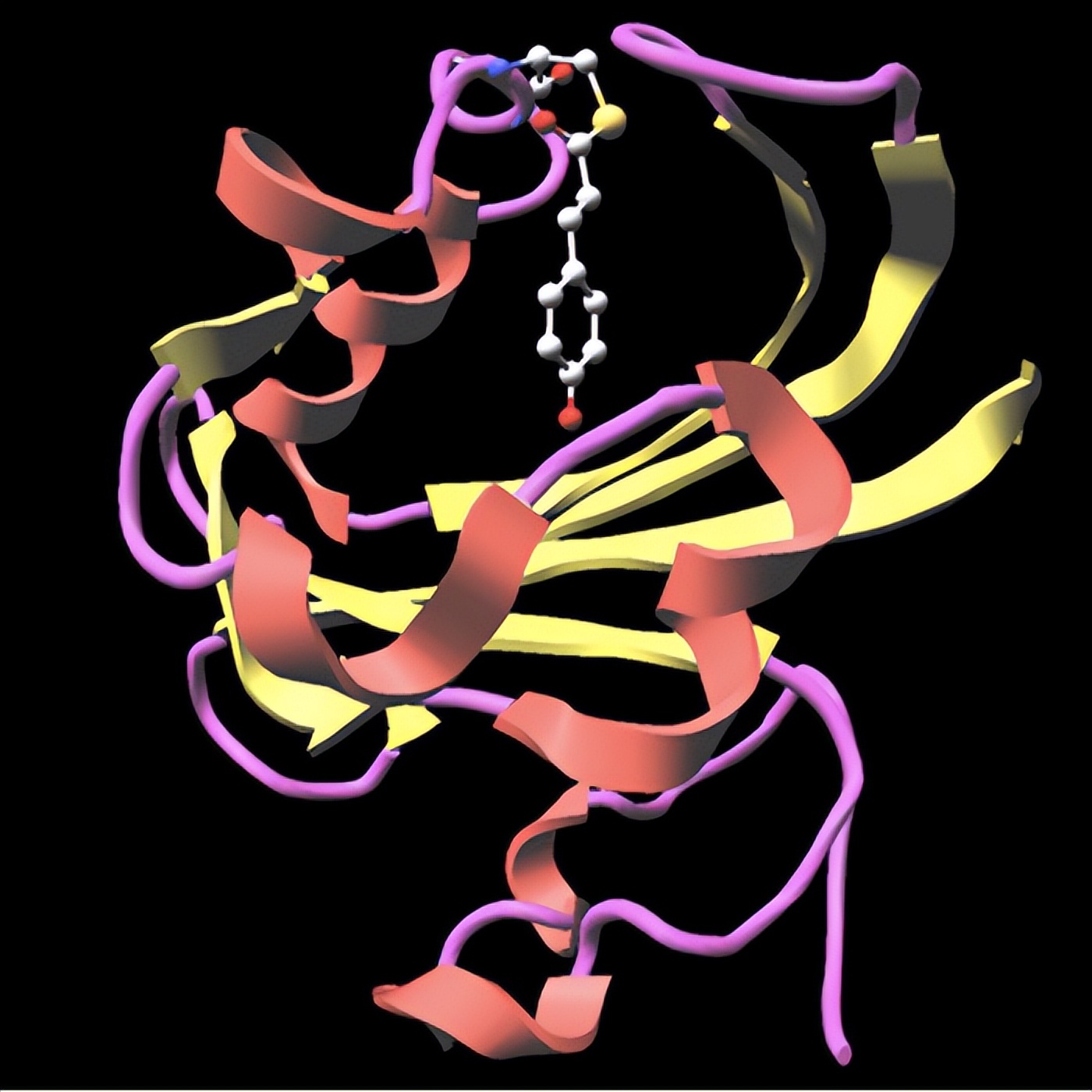
蛋白质及其复杂的结构,几乎是每个生物过程的核心。了解这些分子如何折叠和改变形状是一个重大的科学挑战,因为这是一个与其功能内在相关的过程。传统上,X射线晶体学和核磁共振 (NMR)光谱等工具已被用于破译蛋白质结构。然而,这些方法往往未能捕捉到蛋白质的动态性质,因为它们在生物功能中变形和弯曲。

AlphaFold 2模型是由DeepMind开发的人工智能驱动的工具,它已经通过准确预测蛋白质结构引起了广泛关注。但布朗大学的团队并没有止步于此。他们将AlphaFold 2的能力扩展到静态预测之外,使其能够对蛋白质潜在形状的全系列(the full spectrum of a protein's potential shapes.)进行建模。这一发展标志着向前迈出的关键一步,类 似于捕捉蛋白质运动的高分辨率视频,而不是单一的快照。
用机器学习弥合差距
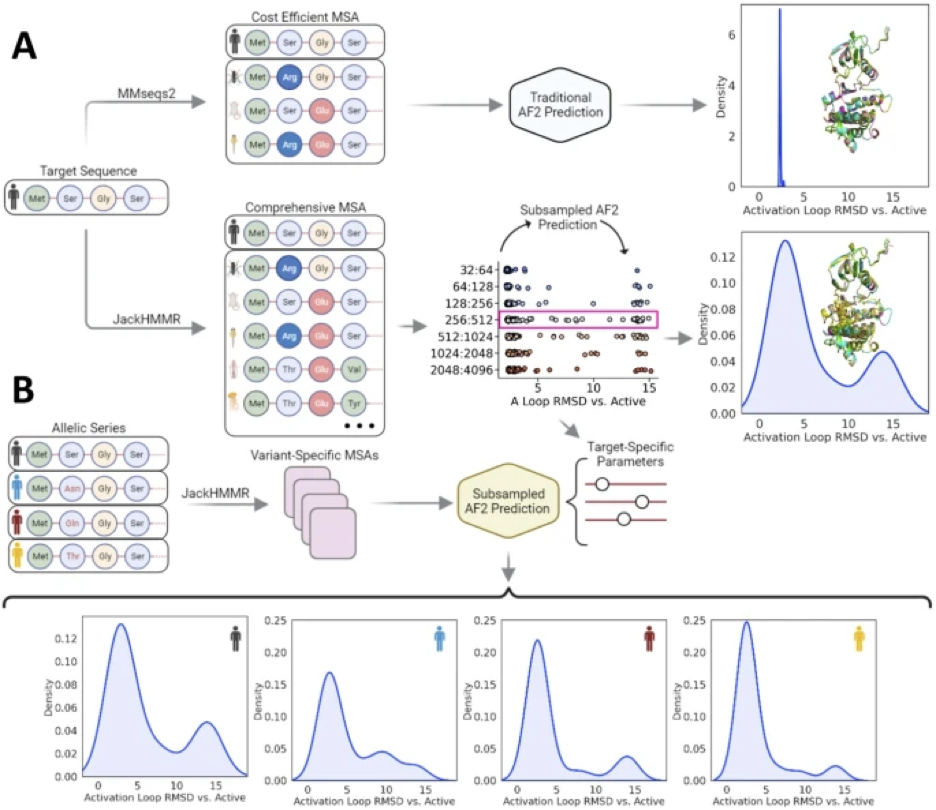
这一突破的关键在于一种名为子采样多序列对齐( subsampling multiple sequence alignments, MSA)的巧妙方法。通过选择性地向AlphaFold 2提供已知蛋白质序列的大量数据库的一部分n内容,研究人员告诉人工智能模型不仅预测一种,而且预测蛋白质可以假设的多种配置。根据核磁共振实验的黄金标准验证,这种方法展示了超过80%的令人印象深刻的准确率,这一壮举充分说明了其潜力。
这种方法的与众不同之处在于其普遍性和效率。在两种蛋白质(Abl1激酶和粒细胞-大噬细胞菌落刺激因子,Abl1 kinase and granulocyte-macrophage colony-stimulating factor(GMCSF))上进行了测试,无论可用序列数据量如何,该方法都被证明是成功的。这种多功能性凸显了其在各种蛋白质中的应用潜力,为药物发现开辟了新的途径,特别是在靶向癌症治疗领域。
从静态到动态:蛋白质建模的新维度

蛋白质从静态建模到动态建模的飞跃不仅仅是技术上的胜利;它代表了科学家如何进行药物发现的范式转变。传统上对蛋白质基态结构的关注限制了研究人员了解蛋白质功能的全部范围的能力,进而广之,药物如何有效地靶向这些分子。通过阐明蛋白质的构象景观,这项研究为设计根据其目标的动态性质进行微调的药物铺平了道路。
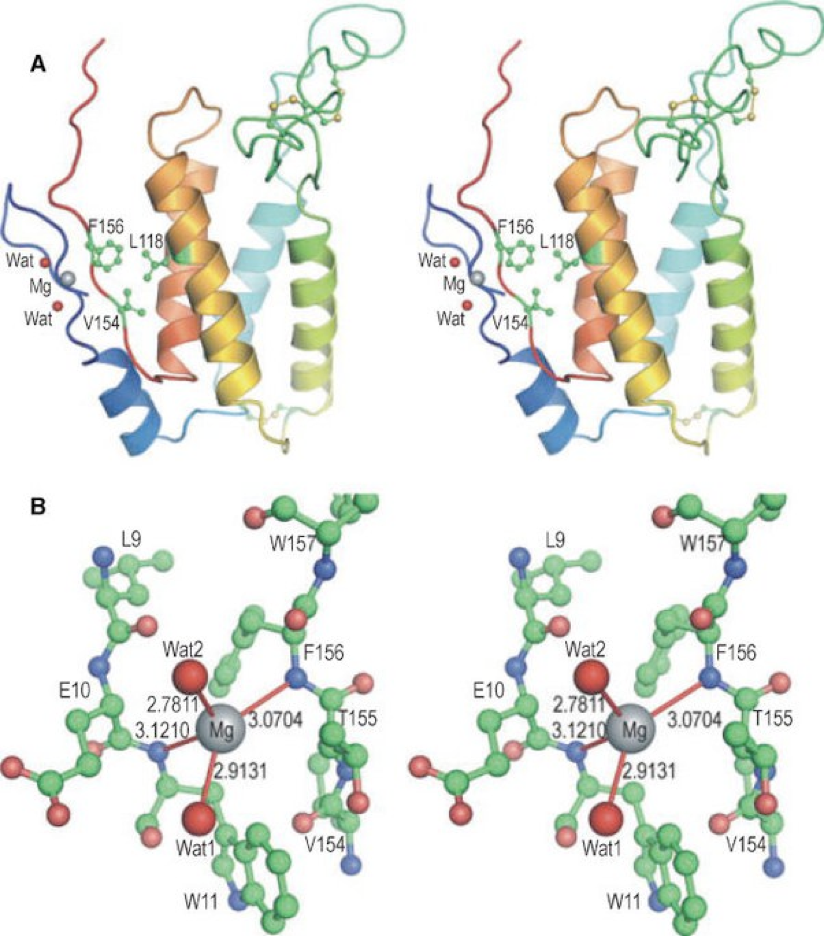
这项研究不仅扩展了我们的科学工具包;他们从根本上丰富了我们对生物过程的理解。他们的工作强调了一个关键的洞察力:要真正掌握药物如何与蛋白质相互作用,必须考虑蛋白质可以采用的各种形状。
影响和未来方向
这项研究的影响是广泛而多样的。例如,它提供了一个新的视角,通过它来看待耐药性的挑战——这是治疗癌症等疾病的一个重要障碍。通过绘制蛋白质结构不断变化的景观,研究人员可以预测突变如何改变药物的功效,指导对耐药性更具弹性的下一代疗法的开发。
此外,这种方法证明了跨学科合作的力量,融合了计算生物学、化学和机器学习领域,以应对医学中一些最紧迫的挑战。它还强调了人工智能和机器学习在科学发现中发挥的日益关键的作用,提供了前所未有的预测能力和效率的工具。

展望未来,布朗大学的团队专注于完善他们的技术,提高其准确性,并扩展其适用性。随着这项研究的不断发展,它有望解锁新的治疗目标,揭示蛋白质动力学的奥秘,并最终迎来药物发现的新时代。
