碳钢–奥氏体不锈钢异种钢焊接如今已广泛应用于各行各业中,特别是在石油运输、海洋勘探等具有腐蚀介质的环境中发挥着重要的作用。由于碳钢和奥氏体不锈钢在热膨胀系数、碳及合金元素含量方面存在较大差别,造成碳钢–奥氏体不锈钢异种钢接头组织和力学性能低于母材,接头处微观组织常常发生元素的偏聚、脆性相的形成以及残余应力的增大等现象,其中接头区域碳偏聚是常见的一种现象[1−2]。通常异种钢焊接过程中,在熔合区各类元素的分布表面上看主要由其浓度差反映,即呈现出梯度分布。然而对于碳钢–奥氏体不锈钢异种钢接头,Pavlovsky[3]等研究发现C元素在碳钢侧熔合区表面发生了偏聚,即形成了一个增碳层,临近区域则出现了贫碳区。进一步针对增、贫碳层研究表明,不锈钢中的Cr元素在焊接过程中向碳钢侧发生了一定的扩散[3−5],增碳层通常含有大量细小板条马氏体、孪晶马氏体、贝氏体和碳的铬化物等混合物,碳元素的偏聚与奥氏体不锈钢中Cr元素扩散进入碳钢熔合区表面与C元素形成稳定的碳化物相关[6]。同时这一增碳层的宽度还与接头高温、长期运行时间密切相关,最终影响到焊接构件的使用寿命。目前异种钢焊接接头处碳迁移微观过程及形成机制,在原子层面还难以解释清楚。
近几年第一性原理的计算方法为金属材料间隙原子扩散机制的研究提供了条件,如使用第一性原理方法探究Cr对Fe–Cr合金、Fe–Cr–Ni合金的影响[7−13],腐蚀介质中Cr元素对合金的影响[14−16]。本文从微观机理研究α-Fe (110)和α-Fe–Cr (110)合金表面C原子的偏聚,在指导实际工程应用中起着举足轻重的作用,为以后在原子和分子角度探究C原子扩散的微观机理提供一个参考。
1. 计算方法和模型第一性原理计算采用CASTEP[17]软件,运用密度泛函理论(DFT)[18−20],结合局域密度近似(LDA)[21]的CA–PZ交换–关联的方法,对α-Fe表面结构中离子和电子的相互作用采用超软赝势(USPP)[22]方法进行描述,其中平面波基组截断能为290 eV,布里渊区积分计算[23]采用Monkhorst–Pack方案,k点网格为4×4×1,迭代过程中,自洽场(SCF)收敛精度为2.0×10–5 eV/atom,原子间最大相互作用力为0.005 eV/nm,原子最大位移变化0.0002 nm。
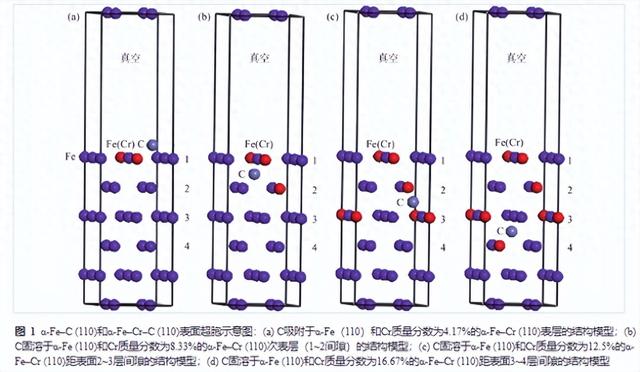
α-Fe (110)、α-Fe–Cr (110)是基于α-Fe块体弛豫优化后得到的表面,本表面模型中根据Fe (110)层结构特点选取6层进行计算。图1是α-Fe (110)、α-Fe–Cr (110)表面超胞的示意图,为了避免平面重叠引起的相互作用,真空层的厚度选取为1 nm。图1(a)为C吸附于α-Fe (110)和Cr质量分数为4.17%的α-Fe–Cr (110)表层的结构模型,图1(b)为C固溶于α-Fe (110)和Cr质量分数为8.33%的α-Fe–Cr (110)次表层(1~2间隙)的结构模型,图1(c)为C固溶于α-Fe (110)和Cr质量分数为12.5%的α-Fe–Cr (110)距表面2~3层间隙的结构模型,图1(d)为C固溶于α-Fe (110)和Cr质量分数为16.67%的α-Fe–Cr (110)距表面3~4层间隙的结构模型。
为了确保计算的可靠性,先对α-Fe块体进行了结构优化,计算的α-Fe块体的晶格参数a和体积v分别为0.2866 nm和0.023551 nm3,与理论值0.2866 nm和0.023541 nm3相比,计算值与理论数值非常接近,因此本计算方法数据可信。
2. 结果和讨论2.1 表面结构的稳定性形成热是原子由单质状态形成化合物时释放的能量,常用于表征化合物形成的难易程度,当形成热为负值时,其绝对值越大,表示该物质越易形成[24−26]。具体到α-Fe–C (110)、α-Fe–Cr (110)和α- Fe–Cr–C (110)表面结构的形成热,可用式(1)计算[27]。

图2所示为碳原子位于α-Fe–C (110)表面结构不同位置时获得的结合能和形成热。结合能曲线来看,碳处于不同位置时,含碳α-Fe–C (110)表面结构的结合能均为负值,即均为稳定结构。具体来看C原子位于α-Fe–C (110)表面层时结合能的值最小,而处于2、3、4层间时,结合能的数值相近且均高于表面为主,表明碳固溶于α-Fe–C (110)表面结构体系中时,C位于表面时对应体系的结合能最小,为最稳定结构。形成热曲线来看,碳所处不同位置时,α-Fe–C (110)表面结构的形成热也均为负值,即碳易固溶于α-Fe–C (110)体系,具体来看C原子位于α-Fe–C (110)表面层时对应的形成热最小,碳更易于在α-Fe–C (110)表面形成。
图3为Cr含量变化对α-Fe–Cr (110)表面结构的形成热和结合能影响曲线。图3(a)结合能曲线可以看出,随着α-Fe–Cr (110)表面结构中Cr含量的增加,α-Fe–Cr (110)表面结构体系的结合能均为负值,即均为稳定结构。具体来看随着Cr含量的增加,α-Fe–Cr (110)体系的结合能数值没有明显的变化,即Cr固溶于α-Fe (110)体系对α-Fe–Cr (110)体系稳定性影响不大。图3(b)形成热曲线来看,Cr含量的增加,使α-Fe–Cr (110)表面结构体系的形成热均为负值,即Cr均可以固溶于α-Fe–Cr (110)体系。具体来看现有的Cr含量研究范围内,Cr含量越高越易于固溶于α-Fe–Cr (110)表面结构体系。
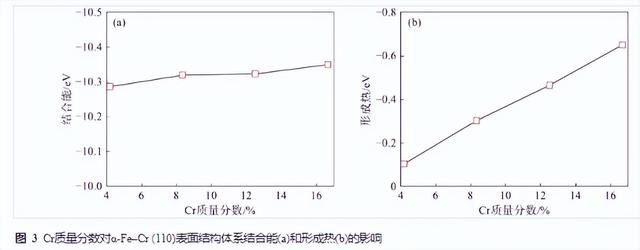
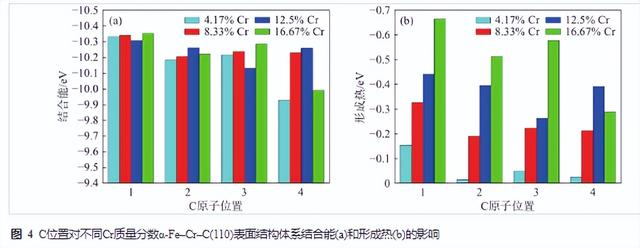
图4为C所处位置对不同Cr含量α-Fe–Cr–C (110)表面结构体系结合能和形成热影响曲线。C溶于不同Cr含量的α-Fe–Cr–C (110)表面结构体系后,结合能的值均为负值,即均为稳定结构。较α-Fe体系相比,Cr含量溶于α-Fe体系后,Cr明显会使α-Fe–Cr–C(110)表面结构体系的结合能数值降低,即提高C在α-Fe–Cr–C (110)体系中的稳定性,对比来看,C处于表面时的稳定性均高于处于次表面和内部位置的2、3、4层间。图4(b)为C处于不同位置时,不同Cr含量α-Fe–Cr–C (110)表面结构体系形成热变化曲线,可以看出Cr含量的增加对含C的α-Fe–Cr–C (110)表面结构体系形成热的影响特别明显,即Cr含量越高,C越有利于溶于α-Fe–Cr–C (110)体系,尤其C分布于α-Fe–Cr–C (110)表面层时的形成热最小,表明C更容易形成于高Cr的α-Fe–Cr表界面并形成稳定的结构。
2.2 碳在α-Fe–Cr(110)体系中偏聚行为表面偏聚能是一个原子吸附于表面位置和溶于内部区域所引起的系统能量之差。针对α-Fe–Cr–C (110)体系来讲,碳在α-Fe–Cr–C (110)表面偏聚能可用式(3)表示[29]:
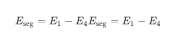
式中E1和E4在α-Fe–C (110)表面分别代表C原子固溶于α-Fe–Cr–C (110)表层和第3~4层间隙的系统总能,在α-Fe–Cr (110)表面分别表示Cr原子取代表面第1层和第4层一个Fe原子的系统总能。Eseg为正说明C原子在表面时能量高,即C原子不易在表面发生偏聚,易于均匀分布于α-Fe–Cr–C (110)体系各个部位,Eseg为负说明C原子在表面时能量低,即C原子易在表面发生偏聚。
图5所示为Cr含量对C原子在α-Fe–Cr–C (110)表面吸附时获得的偏聚能,总体来看C原子固溶于α-Fe (110)、α-Fe–Cr (110)体系时,整个体系的偏聚能均为负值,即C原子均易于在α-Fe (110)、α-Fe–Cr (110)表界面发生聚积。具体到Cr元素的影响来看,α-Fe–Cr–C (110)体系中随着Cr含量的增加有利于减缓C在表界面的偏聚。通过式(3)计算得到,Cr原子在α-Fe–Cr (110)表面偏聚能为–0.613 eV,即Cr原子易于在α-Fe–Cr (110)表面发生聚积。
图6所示为C、Cr原子在碳钢/奥体钢界面偏聚模型,具体到碳钢/奥体钢界面,由于奥氏体钢中Cr含量很高,焊接过程中界面处一定会有一定量的Cr向铁素体中扩散,同时由于碳钢中C含量很高,焊接过程中界面处一定会有一定量的C向奥氏体区域扩散,即在接头界面的碳钢侧出现一个微小α-Fe–C/α-Fe–(Cr)–C界面区域。从以上计算来看,α-Fe–C/α-Fe–(Cr)–C界面靠近碳钢侧,C有向含Cr区域偏聚的倾向,由于Cr的影响,C的偏聚受到了阻碍,使得C更多的偏聚于碳钢/奥体钢界面的碳钢一侧,由于C的偏聚可使得界面结构更加稳定,因此最终在接头焊缝界面的α-Fe–C侧形成了一层高碳的区域。
3. 结论(1)C原子固溶于α-Fe (110)、α-Fe–Cr (110)表面结构体系中,均形成稳定的结构,Cr溶于α-Fe–C (110)表面结构体系中,有利于提高C原子在α-Fe–Cr–C (110)体系中的稳定性。C原子处于α-Fe–Cr–C (110)表面时的稳定性均高于处于次表面和内部。
(2)C原子在α-Fe (110)表面的偏聚倾向高于含铬的α-Fe–Cr (110)表面结构,Cr含量的增加有利于减缓C在表面的偏聚,即C原子更易于偏聚于α-Fe(110)表面而不易扩散于含铬的α-Fe–Cr (110)结构体系中。
文章来源——金属世界
