遗传代谢性肝病(IMLD)是指基因缺陷导致的肝脏代谢异常的一大类疾病,主要表现为肝脏形态结构和(或)功能异常,常伴有其他脏器的损害。

需要指出的是,由于肝脏是机体大多数物质代谢的重要器官,许多遗传代谢性疾病都会累及肝脏。多数情况下,IMLD是单基因遗传性疾病,通常是常染色体隐性遗传,但也可以是常染色体显性遗传、X或Y染色体性连锁遗传等[1]。IMLD起病隐匿,由于该类疾病种类多,临床表现多样且无特异性,这为临床诊断带来了一定困难。
黄疸在临床上较为常见,其中部分病因为相关基因变异,即属于遗传代谢性黄疸。遗传代谢性黄疸的诊断过程对临床医生提出了更高的要求,强调以临床思维为主导。在“医站到底”线上模拟诊疗经验交流会第五期上,首都医科大学附属北京佑安医院郑素军教授分享了3个病例,让我们追随郑教授的病例,一同探索遗传代谢性黄疸的诊疗思路。

病例一

患者基本信息

患者:男,35岁,发现眼黄1年余来诊,无明显不适。
个人史及家族史:否认嗜酒;否认病毒性肝炎等家族史,父母是否黄疸不详。
实验室检查:TBIL:129.6μmol/L ;DBIL:11.3μmol/L ;ALT 25.3U/L ;AST 22.5U/L。WBC 7.47 ×10^9/L,RBC 4.67×10^9/L, HB 152g/L,PLT 163×10^9/L。
乙肝、丙肝、自免肝等相关检查均为阴性结果,甲状腺功能正常。
初步诊断:黄疸原因待查


●思考一:需要进一步做哪些检查来帮助诊断与鉴别诊断?
解答:辅助检查,包括溶血相关检查,肝、胆、脾彩超,基因检测。
♦溶血相关检查:WBC:9.06 ×10^9 /L N 80%;RBC:4.63 × 10^12 /L ,HB 154g/L;PLT:189×10^9/L;网织红细胞绝对值计数570.9×10^9/L(24-84);网织红细胞百分比12.33%(0.5%-1.5%), 均明显升高;RDW: 64.8Fl(40-53);外周血涂片:红细胞轻度大小不等,可见球形红细胞;Coomb’s 试验:阴性。
♦彩超检查:脂肪肝(轻度),脾大(厚48mm,长185mm),肝内低回声(血管瘤?), 胆囊多发结石。
♦UGT1A1基因测序:Crigler- Najjar综合征(包括Ⅰ型和Ⅱ型)常见的32个突变位点检测未发现突变。NGS结果显示ANK1基因杂合突变(c.2467 G>7,p.E823X),为无义突变,导致氨基酸翻译终止,显性遗传(AD)。
●思考二:根据以上信息,如何考虑诊断?如何进行鉴别诊断?诊断思路如何?
解答:
♦诊断:遗传性球型红细胞增多症合并胆石症、脾大。
♦鉴别诊断:Crigler- Najjar综合征(Ⅱ型)
♦诊断思路:诊断UGT1A1相关高间接胆红素血症,需先排除溶血[2]。
♦注意:部分溶血可以没有贫血(本患者即没有贫血),部分可与UGT1A1病共存。

病例二
患者基本信息
患者:女,20岁,皮肤黄染10余年入院,无明显不适。
查体:皮肤、巩膜轻度黄染,余阴性。
个人史及家族史:否认嗜酒、药物应用等;否认病毒性肝炎、肝病等家族史。
实验室检查:TBil:61.6μmol/L;DBil:50.4μmol/L;GGT:18.1U/L;ALP:57.5 U/L;TBA:3.8μmol/L。乙肝、丙肝、自免肝等相关检查均为阴性结果。
初步诊断:黄疸原因待查。
●思考一:初步诊断?
解答:
♦先天性高直接胆红素血症可能性大,考虑是Dubin-johnson综合征或者Rotor综合征。
♦病理检查:轻度非特异性肝炎,结合临床,建议行基因检测,除外Rotor综合征。


♦Rotor综合征应是SLCO1B3、 SLCO1B1双基因纯合突变1,患者只测出来SLCO1B3纯合突变,思路是否有误?

自行设计引物,对SLCO1B1基因进行PCR扩增,产物一代测序(Sanger测序),测出c.1738C>T(p.Arg580Term),纯合变异,为无义突变,导致氨基酸翻译终止!见下。

SLCO1B1-Exon 13-c.1738C>T(p.Arg580Term) : 杂合突变(患者之父)

SLCO1B1-Exon 13-c.1738C>T(p.Arg580Term) : 杂合突变(患者之母)

SLCO1B1-Exon 13-c.1738C>T(p.Arg580Term) :纯合突变(患者)
与NGS测序公司沟通,再次生信分析,找到此纯合突变!先前为漏筛。
故NGS也不完全可靠!一次NGS阴性结果也不要轻易排除诊断。要坚持临床思维为主导,对测序结果有疑问时,应及时与测序公司进行沟通!
最终诊断:支持Rotor综合征
●思考二:Rotor综合征与Dubin-johnson综合征鉴别诊断?
解答:
♦Rotor综合征与Dubin-johnson综合征鉴别诊断[4]。
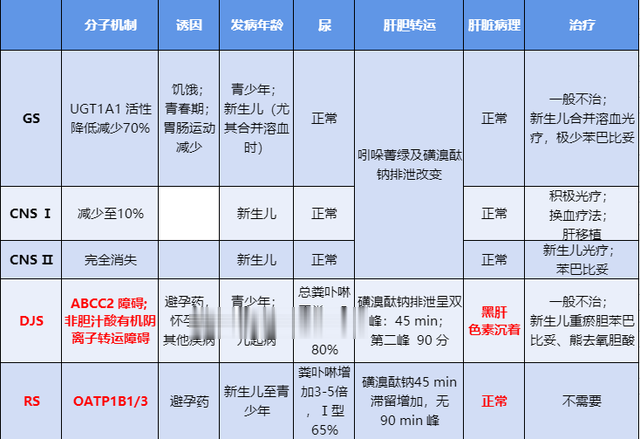
●思考三:先天性高胆红素血症诊断思路?
解答:
♦思路如下:

先天性高胆红素血症的诊断思路[5]
病例三
患者基本信息
患者:男,15岁,因尿黄1年,发现肝硬化2周于2019年8月入院。
查体:皮肤、巩膜中度黄疸。脾脏肋下10cm。
个人史及家族史:否认病毒性肝炎历史,否认腹泻、结肠炎病史,否认药物服用史。
实验室检查:ALT:139.5U/L;AST:183.7U/L;Tbili :107.7μmol/L;Dbil:77.5μmol/L;γ-GT:394.3U/L;Alp:348U/L;TBA:115.5μmol/L;Alb:32.9g/L;WBC:3.41×10^9/L;Hb:121g/L;PLT:77×10^9/L;病毒性标志物、自身免疫性标志物均阴性。
初步诊断:胆汁淤积性肝病。
●思考一:根据以上信息,初步诊断?
解答:胆汁淤积性肝病。

胆汁淤积性肝病诊断流程[6]
♦患者检查发现肝硬化、脾大、肝内外胆管未见明显扩张,食道静脉曲张。通过检查病理提示胆汁淤积性肝硬化:Masson染色显示肝硬化(100x);小叶间胆管减少和细胆管反应(CK7免疫染色);胆栓淤积与Mallory-Denk小体(H&E 400x);肝周细胞出现铜沉积(罗丹宁200x)。
♦基因检测发现患者ABCB4 基因复合杂合变异:新的变异c.T2525C(p.L842P) /c.T3152C(p.V1051A)[7] ,分别来源于母亲、父亲大姐、二姐与患者具有同等情况的复合杂合变异。
♦按照ACMG标准,进行软件预测,为新发现致病变异!
追问家族史:
▪母亲:20多岁曾患胆结石而行胆囊切除术;
▪大姐:曾诊断妊娠期肝内胆汁淤积(ICP);
▪二姐:无症状胆结石( LPAC )。
●思考二:根据以上信息,初步诊断?
解答:胆汁淤积性肝病。
♦诊断:进行性家族性肝内胆汁淤积(PFIC3)[8]
根据家族史和基因型推断家族其他成员患有ABCB4 基因相关性疾病:
▪母亲:低磷脂相关胆石症(LPAC);
▪大姐:妊娠期肝内胆汁淤积(ICP);
▪二姐:低磷脂相关胆石症(LPAC)。
♦鉴别诊断:
▪胆汁酸合成缺陷性疾病:AKR1D1、AMACR、CYP7B1、HSD3B7、CYP7A1、CYP27A1等基因变异;
▪PFIC1、2、4、5、6;
▪胆管细胞分泌障碍:囊性纤维化;
▪胆管发育障碍:Alagille syndrome, polycystic liver diseases, fibropolycystic liver diseases(Caroli disease and congenital hepatic fibrosis);
▪其他:Niemann-Pick病(C1/C2)等。
♦诊断思路:血清GGT水平有助于胆汁淤积鉴别诊断[9]
▪血清GGT:胆道堵塞或梗阻,胆管腔内胆汁酸增多,GGT易从毛细胆管膜洗脱,入血后引起GGT升高。胆汁酸合成、运输障碍(常遗传缺陷),胆管腔内胆汁酸缺少或相对不足,GGT正常或低水平。血清GGT正常的胆汁淤积症,更多见于遗传代谢性疾病。
▪低-GGT胆汁淤积:包括先天性胆汁合成障碍、细胞内胆汁酸转运障碍;PFIC1、2、4、5、6;BRIC1,2,6。
区别:PFIC血清总胆汁酸升高,而胆汁酸合成缺陷患儿总胆汁酸降低或正常。
▪高-GGT胆汁淤积:PFIC3,胆管细胞细胞分泌障碍:囊性纤维化;胆道发育障碍包括Alagille综合征、胆管板发育畸形:polycystic liver diseases, fibropolycystic liver diseases (Caroli disease and congenital hepatic fibrosis)。其他:Niemann-Pick病(C1/C2型)等。
♦遗传性黄疸总诊断思路:

参考文献:
[1]专家访谈:郑素军 遗传代谢性肝病
[2]张杰 ,郑素军. 溶血性黄疸的临床特征及鉴别诊断.临床肝胆病杂志,2020,36(6):1423-1427.
[3]van de Steeg E,et al.Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012 Feb;122(2):519-528.
[4]Wagner KH, et al. Diagnostic criteria and contributors to Gilbert‘s syndrome.Crit Rev Clin Lab Sci. 2018 Mar;55(2):129-139.
[5]白洁,郑素军,段钟平. 4种常见先天性高胆红素血症的临床特征及诊断思路.临床肝胆病杂志,2019,35(8):1680-1683.
[6]中华医学会肝病学分会. 胆汁淤积性肝病管理指南(2021年). 临床肝胆病杂志,2022,38(1):62-69.
[7] Jie Bai, et al. A novel compound heterozygous mutation in ABCB4 gene in a pedigree with progressive familial intrahepatic cholestasis 3: a case report. Annals of translational medicine 2021 Mar;9(5):426 doi:10.21037/atm-20-3747
[8]白洁,郑素军,段钟平.进行性家族性肝内胆汁淤积症的临床特征及诊疗思路。中华肝脏病杂志 ,2021,29(11): 1128-1131.
专家简介

郑素军教授
●郑素军教授
♦医学博士,主任医师,教授,博士研究生导师,首都医科大学附属北京佑安医院肝病中心一科科主任。
♦北京市科技新星,北京市高层次卫生人才,中华医学会肝病学分会肝炎学组委员,中华医学会肝病学分会遗传代谢性肝病协作组委员、秘书,北京医学会肝病委员会委员,北京医学会肝病学分会人工肝与肝衰竭学组委员,中国研究型医院学会肝病专委会肝纤维化学组委员。
♦《World Journal of Gastroenterology》、《临床肝胆病杂志》、《胃肠病学与肝病学杂志》等编委,曾先后独立承担国家自然科学基金、北京市自然科学基金、首都医学发展重点攻关专项基金等10余项,近年发表通讯或第一作者SCI收录28篇。
来源:肝胆相照平台